Does the newer version of Trinity (v2.8.4) make better transcriptome assemblies (for killifish) than the old (v2.2.0)?
tl;dr Yes.
XSEDE Computing Resources
Since I'm overdrawn on my new XSEDE allocation on the PSC Bridges LM (large memory) partition after receiving it on 11/26/2018, a report is required to apply for more.
In case you couldn't tell, I'm a really big fan of the PSC Bridges hpc! I've used 1862 SU out of the 1000 SU they allocated to me on the LM partition and 11484 out of 13000 SU on the RM (regular memory).
In case it might benefit others, here is roughly the progress report that I will submit with my request.
Re-assembling 17 Fundulus killifish transcriptomes
Since receiving my allocation to the PSC Bridges LM partition on 11/26/2018, I have re-assembled 17 transcriptomes from 16 species of Fundulus killifish using the Trinity de novo transcriptome assembler version 2.8.4. I originally assembled transcriptomes from the 16 Fundulus species using Trinity version 2.2.0.
There is some evidence that assemblies can be improved by using the updated versions of Trinity. I wanted to re-assemble these killifish transcriptomes to see if the assemblies could be improved.
These transcriptomes will be used for the purpose of studying the history of adaptation to different salinities. Some Fundulus species can tolerate a range of salinities (euryhaline) by switching osmoregulatory mechanisms while others require a more narrow salinity range (stenohaline) in either fresh or marine waters. This large set of RNAseq data from multiple species is going to help us better characterize the divergence of these molecular osmoregulatory mechanisms between euryhaline and stenohaline freshwater species. To that end, I am attempting to develop a pipeline for orthologous gene expression profiling analysis across species.
This profiling analysis will enable us to compare expression patterns of salinity-responsive genes, e.g. aquaporin 3 (shown below) across clades of species.
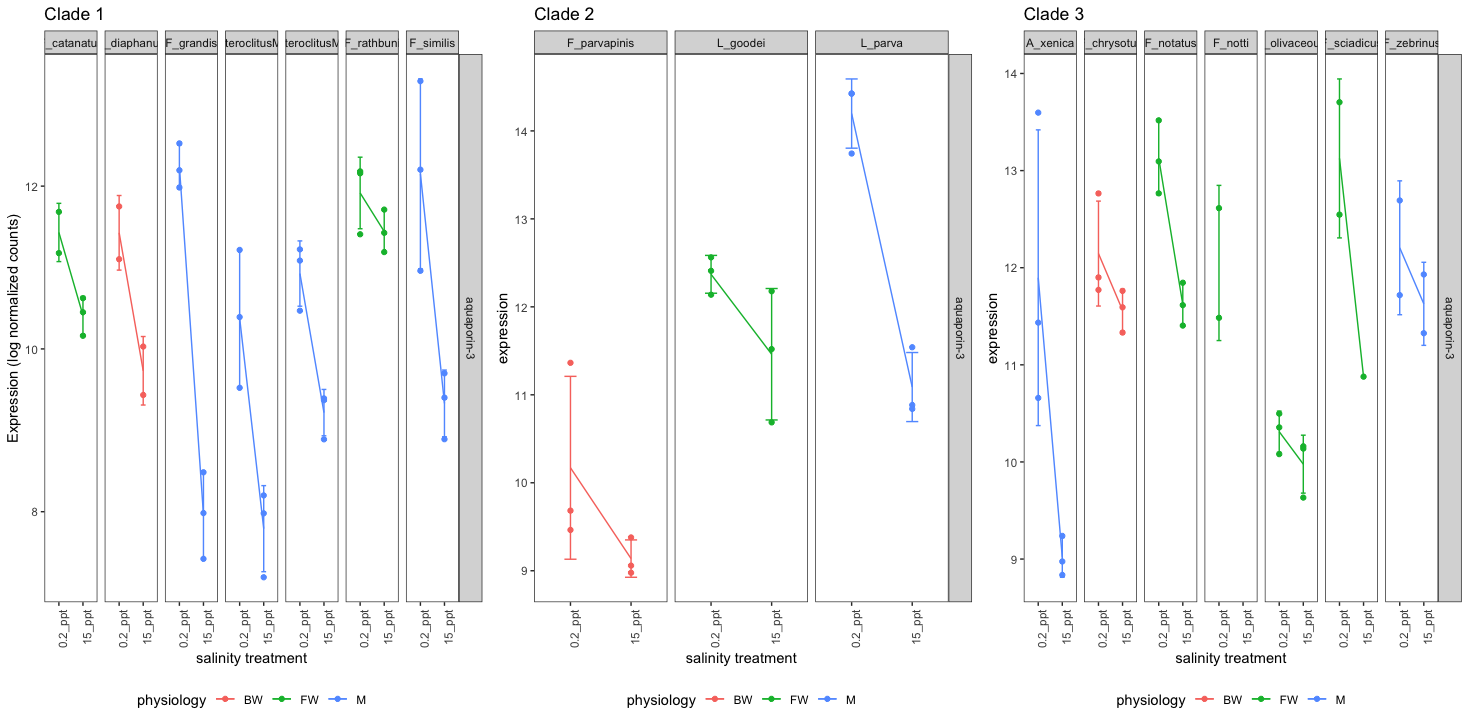
The nature of the fragmented assembly output from Trinity makes it especially challenging in our attempt to develop this pipeline. We want to make sure that the reference transcriptomes are the best that they can be so that expression profiles are accurately reflected across species.
Are there differences between the v2.2.0 and v2.8.4 assemblies?
Yes. Are they better? It seems so. But, there are a few items to investigate further.
Below are comparisons of various evaluation metrics between the 17 old v2.2.0 (blue) and new v2.8.4 (green) Trinity assemblies. On the left are slope graphs comparing the metric between both assemblies and on the right are split violin plots showing the distributions of all the assemblies.
Shout out to Dr. Harriet Alexander for helping with the code to visualize these types of comparisons for our paper comparing re-assemblies for the MMETSP - coming out soon in GigaScience!
Code for making these plots is here.
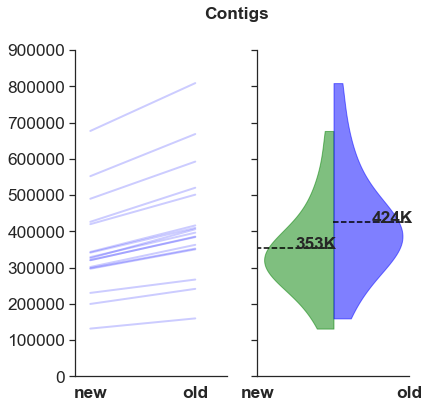
There are fewer contigs.
However, the large numbers >100,000 contigs indicates that these assemblies are still very fragmented. Orthogroup/ortholog prediction is required for downstream analysis.
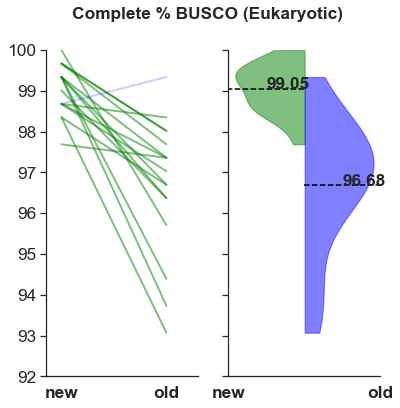
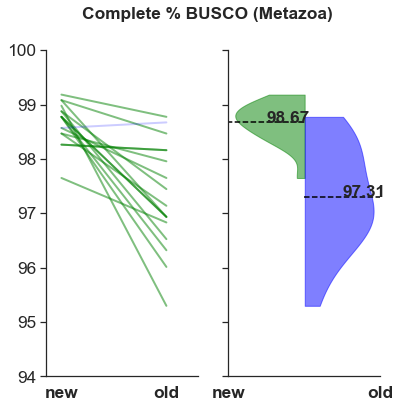
The BUSCO scores are higher.
What makes these assemblies especially appealing is their higher scores with the Benchmarking Universal Single-Copy Ortholog (BUSCO) assessment tool. One species, Lucania goodei had 100%! The distribution of scores is tighter for the new assemblies compared to the old.
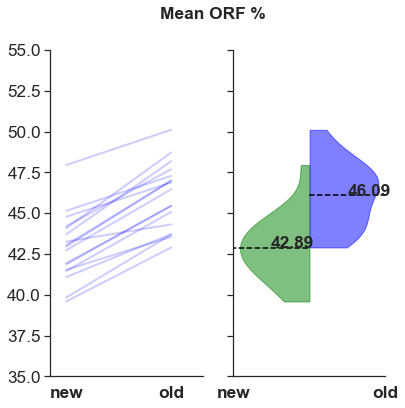
Lower % ORF?
For some reason, mean % ORF decreases. This was not expected.
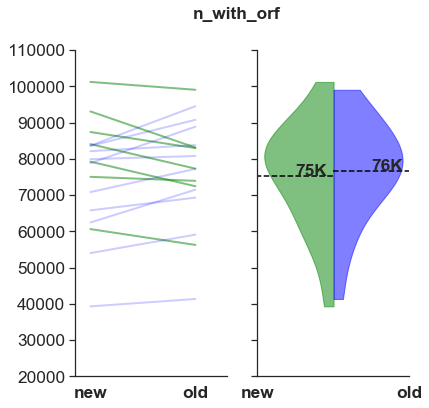
Number of contigs with ORF
This is roughly the same and a very low number.
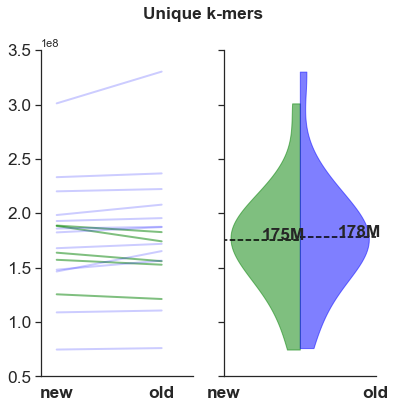
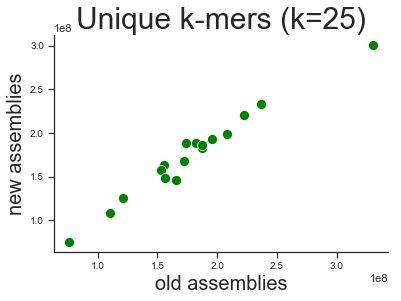
Similar unique k-mers
The number of unique k-mers (k=25) does not really change. This means that the content is similar.
Conditional Reciprocal Best Blast
Using transrate --assembly reference mode to examine the comparative metrics with Conditional Reciprocal Best BLAST (CRBB) between assemblies consistently did not work, for some reason. (Whereas it did work for comparison with the NCBI version of the Fundulus heteroclitus sister species) This requires further investigation.
Version:
Transrate v1.0.3
by Richard Smith-Unna, Chris Boursnell, Rob Patro,
Julian Hibberd, and Steve Kelly
Command:
transrate --assembly=/pylon5/bi5fpmp/ljcohen/kfish_trinity/F_parvapinis.trinity_out.Trinity.fasta --reference=/pylon5/bi5fpmp/ljcohen/kfish_assemblies_old/F_parvapinis.trinity_out.Trinity.fasta --threads=8 --output=/pylon5/bi5fpmp/ljcohen/kfish_transrate/F_parvapinis_trinity_v_old/
Output:
[ INFO] 2018-12-06 22:23:47 : Loading assembly: /pylon5/bi5fpmp/ljcohen/kfish_trinity/F_parvapinis.trinity_out.Trinity.fasta
[ INFO] 2018-12-06 22:24:45 : Analysing assembly: /pylon5/bi5fpmp/ljcohen/kfish_trinity/F_parvapinis.trinity_out.Trinity.fasta
[ INFO] 2018-12-06 22:24:45 : Results will be saved in /pylon5/bi5fpmp/ljcohen/kfish_transrate/F_parvapinis_trinity_v_old/F_parvapinis.trinity_out.Trinity
[ INFO] 2018-12-06 22:24:45 : Calculating contig metrics...
[ INFO] 2018-12-06 22:25:37 : Contig metrics:
[ INFO] 2018-12-06 22:25:37 : -----------------------------------
[ INFO] 2018-12-06 22:25:37 : n seqs 298549
[ INFO] 2018-12-06 22:25:37 : smallest 183
[ INFO] 2018-12-06 22:25:37 : largest 27771
[ INFO] 2018-12-06 22:25:37 : n bases 310786992
[ INFO] 2018-12-06 22:25:37 : mean len 1040.98
[ INFO] 2018-12-06 22:25:37 : n under 200 15
[ INFO] 2018-12-06 22:25:37 : n over 1k 77121
[ INFO] 2018-12-06 22:25:37 : n over 10k 802
[ INFO] 2018-12-06 22:25:37 : n with orf 62453
[ INFO] 2018-12-06 22:25:37 : mean orf percent 43.26
[ INFO] 2018-12-06 22:25:37 : n90 340
[ INFO] 2018-12-06 22:25:37 : n70 1141
[ INFO] 2018-12-06 22:25:37 : n50 2512
[ INFO] 2018-12-06 22:25:37 : n30 4111
[ INFO] 2018-12-06 22:25:37 : n10 6966
[ INFO] 2018-12-06 22:25:37 : gc 0.46
[ INFO] 2018-12-06 22:25:37 : bases n 0
[ INFO] 2018-12-06 22:25:37 : proportion n 0.0
[ INFO] 2018-12-06 22:25:37 : Contig metrics done in 52 seconds
[ INFO] 2018-12-06 22:25:37 : No reads provided, skipping read diagnostics
[ INFO] 2018-12-06 22:25:37 : Calculating comparative metrics...
[ INFO] 2018-12-06 23:23:24 : Comparative metrics:
[ INFO] 2018-12-06 23:23:24 : -----------------------------------
[ INFO] 2018-12-06 23:23:24 : CRBB hits 0
[ INFO] 2018-12-06 23:23:24 : n contigs with CRBB 0
[ INFO] 2018-12-06 23:23:24 : p contigs with CRBB 0.0
[ INFO] 2018-12-06 23:23:24 : rbh per reference 0.0
[ INFO] 2018-12-06 23:23:24 : n refs with CRBB 0
[ INFO] 2018-12-06 23:23:24 : p refs with CRBB 0.0
[ INFO] 2018-12-06 23:23:24 : cov25 0
[ INFO] 2018-12-06 23:23:24 : p cov25 0.0
[ INFO] 2018-12-06 23:23:24 : cov50 0
[ INFO] 2018-12-06 23:23:24 : p cov50 0.0
[ INFO] 2018-12-06 23:23:24 : cov75 0
[ INFO] 2018-12-06 23:23:24 : p cov75 0.0
[ INFO] 2018-12-06 23:23:24 : cov85 0
[ INFO] 2018-12-06 23:23:24 : p cov85 0.0
[ INFO] 2018-12-06 23:23:24 : cov95 0
[ INFO] 2018-12-06 23:23:24 : p cov95 0.0
[ INFO] 2018-12-06 23:23:24 : reference coverage 0.0
[ INFO] 2018-12-06 23:23:24 : Comparative metrics done in 3467 seconds
[ INFO] 2018-12-06 23:23:24 : -----------------------------------
[ INFO] 2018-12-06 23:23:24 : Writing contig metrics for each contig to /pylon5/bi5fpmp/ljcohen/kfish_transrate/F_parvapinis_trinity_v_old/F_parvapinis.trinity_out.Trinity/contigs.csv
[ INFO] 2018-12-06 23:23:43 : Writing analysis results to assemblies.csv
Conclusion
Trinity v2.8.4 is better than v2.2.0. While v2.8.4 still produces very fragmented assemblies, the higher BUSCO content is exciting.
There are questions requiring further investigation
- Why, if the unique k-mer content is similiar, would the BUSCO scores improve between versions?
- ORF content (number of contigs with ORF and mean ORF %) are parodoxical. Why would the ORF content decrease in the newer assemblies?
- Why wouldn't transrate
---referencework to get CRBB metrics between these assemblies?
What has improved in Trinity v2.8.4?
According to the release notes from Trinity, major improvements have included using salmon expression quantification to help with filtering out assembly artifacts and overhauling the "supertranscript module" to deal with high polymorphism situations. Since killifish are highly heterozygous, we are likely benefitting from these improvements.
Future directions
In addition to annotating these transcritomes and continuing to investigate the questions above, I would also like to try Matt MacManes' Oyster River Protocol and accompanying 'orthofuser' script to combine contigs from different assemblers.
Comments
comments powered by Disqus